211service.com
Första atomnivåsimuleringen av ett helt batteri
När det gäller att utveckla nästa generations teknik är den största flaskhalsen utan tvekan batteriet. Ingenjörer behöver bättre batterier för elfordon, för energilagring i elnät och, naturligtvis, för konsumentelektronik.
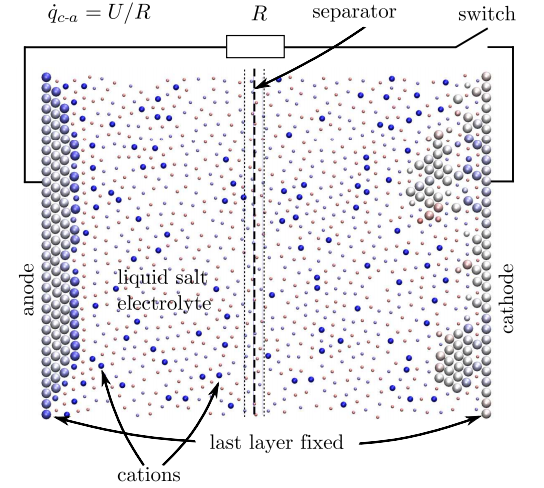
Dessa batterier måste leverera en högre ström över fler urladdningscykler med en högre energitäthet, för att bara nämna några av utmaningarna.
Att bygga och testa nya batteridesigner är tidskrävande, svårt och dyrt. Så det är praktiskt för elektrokemister att simulera hur ett batteri fungerar innan de någonsin blir smutsiga.
Det är knepigt. Ingen har kunnat simulera ett helt batteri på atomnivå på grund av komplexiteten i de processer som pågår och begränsningarna i dagens modelleringstekniker.
Idag förändras det tack vare arbetet av Wolf Dapp vid Institutet för avancerad simulering och Martin Muser vid universitetet i Saarlandes, båda i Tyskland. De här killarna har simulerat beteendet hos ett helt batteri på atomär skala. Och deras simulering återger många av de verkliga egenskaperna hos ett batteri från första principer för första gången.
Under de senaste åren har datavetare gjort betydande framsteg när det gäller att simulera olika aspekter av batteribeteende. Dessa modeller fungerar på mesoskalan - mindre än elektroderna men större än molekylerna. Simuleringarna är beroende av experimentella data för att modellera saker som jonisk och elektronisk konduktivitet, diffusionskoefficienter, strömtätheter, elektrokemiska potentialer och så vidare.
Dessa modeller har en allvarlig nackdel – de har liten prediktiv kraft när det kommer till nya material eller kombinationer av material för vilka experimentella data inte finns tillgängliga. För att förutsäga beteendet hos nya material måste elektrokemister modellera batterier i skalan av atomer och molekyler.
Det är svårt eftersom de tekniker som datavetare använder för att simulera beteendet hos atomer och molekyler inte är lämpliga för batterier. Dessa simuleringar är designade för system som är i jämvikt eller nära det. De fungerar genom att utjämna den kemiska potentialen eller minimera energin i systemet. Men skillnaden i den kemiska potentialen mellan två elektroder är just det som driver laddningstransporten i ett batteri, säger Dapp och Muser.
Så för att modellera ett batteri som helhet måste datormodellen ta hänsyn till varje förändring i energi eller kemisk potential vid varje steg i beräkningen. Detta är precis vad Dapp och Muser har gjort. I deras modell är laddning en variabel som kan utbytas mellan atomer och över bindningar i varje steg av beräkningen.
De resulterande simuleringarna är små men imponerande. Deras nanobatteri består av 358 atomer, varav 118 utgör elektroderna. Katoden täcks initialt med ett skikt av 20 atomer med 39 positivt joniserade atomer lösta i elektrolyten.
Beräkningen fortsätter sedan i steg där atomer kan röra sig och utbyta laddning allteftersom systemet utvecklas. Hela simuleringen består av cirka 10 miljoner av dessa steg.
Resultaten är anmärkningsvärda eftersom de faktiskt återger de generiska urladdningskurvorna för riktiga makroskopiska batterier. Till exempel minskar en lägre driftstemperatur det simulerade batteriets effektiva kapacitet. Och viktigast av allt, simuleringen återger hur vanliga batterier slits ut. Vid omladdning försämras batteriets prestanda något, och elektrodens ytmorfologi förändras under batteriets drift, säger Dapp och Muser.
Dessa killar säger att arbetet i det här skedet bara är en proof-of-principe-modell och att det finns olika sätt på vilka det kan förbättras i framtiden. Till exempel modellerar de elektrolyten med hjälp av partiklar som har en fast laddning och därför inte kan byta ut den.
Det är inte så elektrolyter fungerar i riktiga batterier och så detta är en viktig brist i den nya metoden. Men Dapp och Muser tänker rätta till detta. Denna idealisering kommer att överges i framtida arbete, säger de.
Sammantaget ser detta ut att vara ett viktigt arbete. Denna typ av modell kan dramatiskt förbättra prediktiva kraften i batterisimuleringar och därmed bidra till att spara elektrokemister avsevärd tid, energi och pengar innan de påbörjar detaljerade experiment.
Slutresultatet borde bli bättre batterier men det är en lång väg kvar innan dess.
Ref: arxiv.org/abs/1308.3424 : Redoxreaktioner med empiriska möjligheter: Atomistiska batteriurladdningssimuleringar