211service.com
De oseriösa immuncellerna som förstör hjärnan
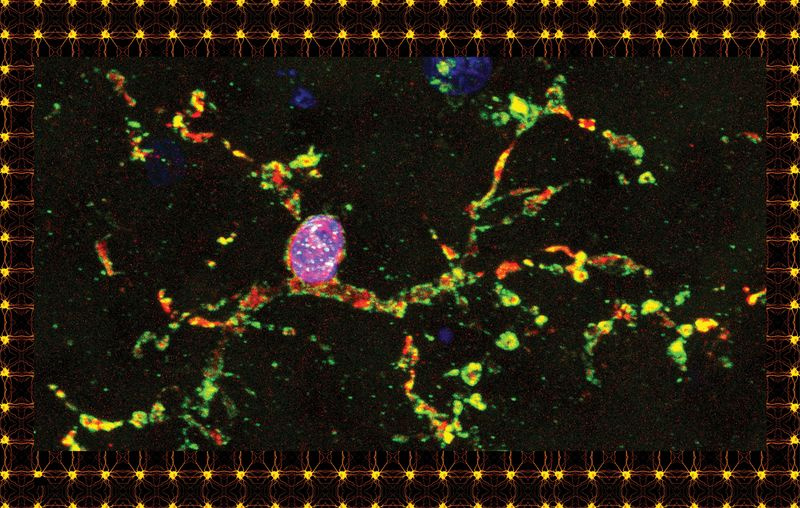
Under de första åren av sin karriär inom hjärnforskning tänkte Beth Stevens på mikroglia med irritation om hon överhuvudtaget tänkte på dem. När hon tittade in i ett mikroskop och såg dessa allestädes närvarande celler med sina spindelformade tentakler, gjorde hon det som de flesta neuroforskare hade gjort i generationer: hon tittade rakt förbi dem och fokuserade på resten av hjärnvävnaden, precis som du kan se genom prickar av smuts på en vindruta.
Vad gör de där? hon trodde. De är i vägen.'

Den här historien var en del av vårt majnummer 2016
- Se resten av frågan
- Prenumerera
Stevens skulle aldrig ha gissat att hon bara några år senare skulle köra en laboratorium vid Harvard och Bostons barnsjukhus ägnade åt studiet av dessa obskyra små klumpar. Eller att hon skulle hävda i världens främsta vetenskapliga tidskrifter att mikroglia kan ha nyckeln till att förstå inte bara normal hjärnutveckling utan också vad som orsakar Alzheimers, Huntingtons, autism, schizofreni och andra svårlösta hjärnsjukdomar.
Mikroglia är en del av en större klass av celler - gemensamt känd som glia - som utför en rad funktioner i hjärnan, styr dess utveckling och fungerar som dess immunsystem genom att sluka upp sjuka eller skadade celler och transportera bort skräp. Tillsammans med sin frekventa medarbetare och mentor, Stanford-biologen Ben Barres, och en växande kader av andra forskare, visar Stevens, 45, att dessa länge förbisedda celler är mer än bara stödarbetare för nervcellerna de omger. Hennes arbete har väckt ett provokativt förslag: att hjärnsjukdomar på något sätt kan utlösas av att vårt eget kroppsliga försvar blivit dåligt.
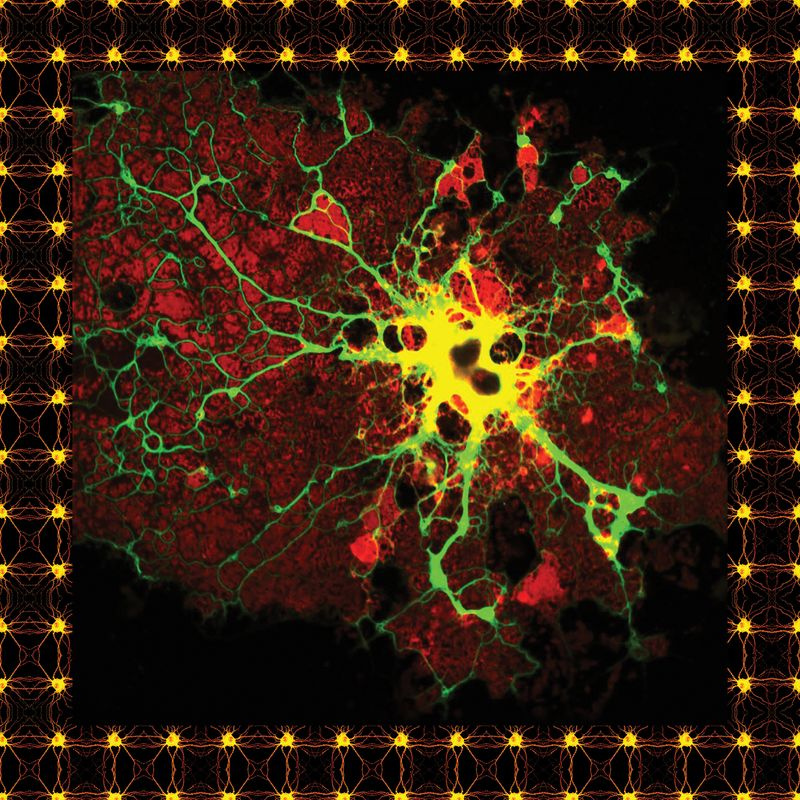
Överst på sidan: En mikrogliacell från en mänsklig hjärna, färgad för forskningsändamål. Ovan: En typ av gliacell känd som en oligodendrocyt.
I ett banbrytande papper , i januari visade Stevens och forskare vid Broad Institute of MIT och Harvard att avvikande mikroglia kan spela en roll vid schizofreni - orsakar eller åtminstone bidrar till den massiva cellförlusten som kan lämna människor med förödande kognitiva defekter. Av avgörande betydelse pekade forskarna på en kemisk väg som kan vara inriktad på att bromsa eller stoppa sjukdomen. Förra veckan, Stevens och andra forskare publiceras ett liknande fynd för Alzheimers.
Det här kanske bara är början. Stevens undersöker också sambandet mellan dessa små strukturer och andra neurologiska sjukdomar - arbete som gav henne 625 000 USD MacArthur Foundation genistipendium september förra året.
Allt detta väcker spännande frågor. Är det möjligt att många vanliga hjärnsjukdomar, trots sina vittgående symtom, orsakas eller åtminstone förvärras av samma gärningsman, en del av immunsystemet? Om så är fallet, skulle många av dessa störningar kunna behandlas på liknande sätt – genom att stoppa dessa oseriösa celler?
Komplexa maskiner
Det är inte förvånande att forskare i flera år har ignorerat mikroglia och andra gliaceller till förmån för neuroner. Neuroner som eldar tillsammans gör att vi kan tänka, andas och röra oss. Vi ser, hör och känner med hjälp av neuroner, och vi bildar minnen och associationer när kopplingarna mellan olika neuroner stärks i förbindelserna mellan dem, så kallade synapser. Många neuroforskare hävdar att nervceller skapar vårt medvetande.
Glia, å andra sidan, har alltid ansetts vara mindre viktig och intressant. De har fotgängaruppgifter som att tillföra näringsämnen och syre till nervceller, samt att torka upp herrelösa kemikalier och transportera bort sopor.
Forskare har känt till glia under en tid. På 1800-talet noterade patologen Rudolf Virchow förekomsten av små runda celler som packar utrymmena mellan neuroner och kallade dem nervenkitt eller neuroglia, som kan översättas som nervkitt eller lim. En variant av dessa celler, känd som astrocyter, definierades 1893. Och sedan på 1920-talet utvecklade den spanska vetenskapsmannen Pio del Río Hortega nya sätt att färga celler som tagits från hjärnan. Detta fick honom att identifiera och namnge ytterligare två typer av gliaceller, inklusive mikroglia, som är mycket mindre än de andra och kännetecknas av sin spindelform och flera grenar. Det är först när hjärnan skadas i vuxen ålder, föreslog han, som mikroglia vaknar till liv - rusar till skadan, där man trodde att de hjälpte till att städa upp området genom att äta skadade och döda celler. Astrocyter dök också ofta upp på scenen; man trodde att de skapade ärrvävnad.
Denna nödkonvergens av mikroglia och astrocyter kallades glios, och när Ben Barres började på läkarutbildningen i slutet av 1970-talet var den väletablerad som ett kännetecken för neurodegenerativa sjukdomar, infektioner och ett brett spektrum av andra medicinska tillstånd. Men ingen verkade förstå varför det hände. Det fascinerade Barres, då neurolog under utbildning, som såg det varje gång han tittade i mikroskop på nervvävnad i nöd. Det var bara riktigt fascinerande, säger han. Det stora mysteriet var: vad är poängen med denna glios? Är det bra? Är det dåligt? Driver det sjukdomsprocessen, eller försöker den reparera den skadade hjärnan?
Barres började leta efter svaret. Han lärde sig hur man odlar gliaceller i en skål och tillämpar en ny inspelningsteknik på dem. Han kunde mäta deras elektriska egenskaper, som bestämmer den biokemiska signalering som alla hjärnceller använder för att kommunicera och koordinera aktivitet.
Från den andra gången jag började spela in gliacellerna tänkte jag 'Herregud!' minns Barres. Den elektriska aktiviteten var mer dynamisk och komplex än någon hade trott. Dessa märkliga elektriska egenskaper kunde endast förklaras om gliacellerna var anpassade till förhållandena runt dem och till de signaler som frigörs från närliggande neuroner. Barres gliaceller hade med andra ord alla maskiner som var nödvändiga för att engagera sig i en komplex dialog med neuroner, och förmodligen för att svara på olika typer av tillstånd i hjärnan.
Varför skulle de dock behöva detta maskineri om de bara var involverade i att städa upp döda celler? Vad skulle de kunna göra? Det visar sig att i frånvaro av kemikalier som frigörs av glia, begick neuronerna den biokemiska versionen av självmord. Barres visade också att astrocyterna verkade spela en avgörande roll för att bilda synapser, de mikroskopiska kopplingarna mellan neuroner som kodar minnet. I isolering kunde neuroner bilda de taggiga bihangen som var nödvändiga för att nå synapserna. Men utan astrocyter var de oförmögna att ansluta till varandra.
Knappt någon trodde honom. När han var en ung fakultetsmedlem vid Stanford på 1990-talet avslogs en av hans anslagsansökningar till National Institutes of Health sju gånger. Recensenter sa hela tiden: 'Nä, det finns inget sätt att glia kan göra det här', minns Barres. Och även efter att vi publicerat två tidningar i Vetenskap Jag visade att [astrocyter] hade djupgående, nästan allt-eller-inget-effekter för att kontrollera synapsernas bildning eller synapsaktivitet, jag kunde fortfarande inte få pengar! Jag tror att det fortfarande är svårt att få folk att tänka på glia som att göra något aktivt i nervsystemet.
Märkt för eliminering
Beth Stevens kom för att studera glia av en slump. Efter examen från Northeastern University 1993 följde hon sin blivande make till Washington, D.C., där han hade fått arbete i den amerikanska senaten. Stevens hade förbehandlats på college och hoppades få arbeta i ett labb vid National Institutes of Health. Men utan tidigare forskningserfarenhet fick hon ett rejält avslag. Så hon tog ett jobb som väntade bord på en Chili's-restaurang i närliggande Rockville, Maryland, och dök upp på NIH med sitt CV varje vecka.
Efter några månader fick Stevens ett samtal från en forskare vid namn Doug Fields, som behövde hjälp i sitt labb. Fields studerade krångligheterna i processen där nervceller isoleras i en beläggning som kallas myelin. Denna isolering är avgörande för överföringen av elektriska impulser.
När Stevens tillbringade de följande åren med att doktorera vid University of Maryland, blev hon fascinerad av den roll som gliaceller spelade för att isolera neuroner. Längs vägen blev hon bekant med andra insikter om gliaceller som började dyka upp, särskilt från Ben Barres labb. Det är därför som Stevens, strax efter att ha avslutat sin doktorsexamen 2003, fann sig själv som postdoc i Barres labb i Stanford, på väg att göra en avgörande upptäckt.
Barres grupp hade börjat identifiera de specifika föreningar som astrocyter utsöndrade som verkade få neuroner att växa synapser. Och så småningom märkte de att dessa föreningar också stimulerade produktionen av ett protein som heter C1q.
Konventionell visdom ansåg att C1q endast aktiverades i sjuka celler - proteinet markerade att de skulle ätas upp av immunceller - och endast utanför hjärnan. Men Barres hade hittat det i hjärnan. Och det var i friska neuroner som utan tvekan var på sitt mest robusta stadium: i tidig utveckling. Vad gjorde C1q-proteinet där?
En färgad astrocyt.
Svaret ligger i det faktum att markering av celler för eliminering inte är något som bara sker i sjuka hjärnor; det är också viktigt för utvecklingen. När hjärnor utvecklas bildar deras neuroner mycket mer synaptiska kopplingar än vad de så småningom kommer att behöva. Endast de som används får vara kvar. Denna beskärning möjliggör det mest effektiva flödet av neurala överföringar i hjärnan, och tar bort brus som kan smutsa ner signalen.
Men det var okänt hur exakt processen fungerade. Var det möjligt att C1q hjälpte till att signalera hjärnan att beskära oanvända synapser? Stevens fokuserade sin postdoktorala forskning på att ta reda på det. Vi kunde ha haft helt fel, minns hon. Men vi gick för det.
Det lönade sig. I ett papper från 2007 visade Barres och Stevens att C1q verkligen spelar en roll för att eliminera onödiga neuroner i den utvecklande hjärnan. Och de fann att proteinet är praktiskt taget frånvarande i friska vuxna neuroner.
Nu stod forskarna inför ett nytt pussel. Visas C1q i hjärnsjukdomar eftersom samma mekanism som är involverad i beskärning av en utvecklande hjärna senare går snett? Faktum är att bevis redan växte på att en av de tidigaste händelserna i neurodegenerativa sjukdomar som Alzheimers, Parkinsons och Huntingtons var betydande förlust av synapser.
Vi kanske äntligen går efter sjukdomar som har löpt okontrollerat i generationer.
När Stevens och Barres undersökte möss som fötts upp för att utveckla glaukom, en neurodegenerativ sjukdom som dödar nervceller i det optiska systemet, fann de att C1q dök upp långt innan något annat detekterbart tecken på att sjukdomen tog fäste. Det dök upp redan innan cellerna började dö.
Detta antydde att immuncellerna faktiskt kan orsaka sjukdomen, eller åtminstone påskynda den. Och det erbjöd en spännande möjlighet: att något kunde göras för att stoppa processen. Barres grundade ett företag, Annexon Biosciences, för att utveckla läkemedel som kunde blockera C1q. Förra veckans papper publicerad av Barres, Stevens och andra forskare visar att en förening som testas av Annexon verkar kunna förhindra uppkomsten av Alzheimers hos möss som fötts upp för att utveckla sjukdomen. Nu hoppas företaget kunna testa det på människor under de kommande två åren.
Vägar till behandlingar
För att bättre förstå processen som C1q hjälper till att utlösa, ville Stevens och Barres ta reda på vad som faktiskt spelar rollen som Pac-Man, som äter upp synapserna som är markerade för döden. Det var välkänt att vita blodkroppar kända som makrofager slukade upp sjuka celler och främmande inkräktare i resten av kroppen. Men makrofager finns vanligtvis inte i hjärnan. För att deras teori skulle fungera måste det finnas någon annan mekanism. Och ytterligare forskning har visat att cellerna som äter även i friska hjärnor är dessa mystiska kluster av material som Beth Stevens i flera år hade tittat förbi i mikroskopet – mikroglian som Río Hortega identifierade för nästan 100 år sedan.
Nu ägnar Stevens labb vid Harvard, som hon öppnade 2008, hälften av sina ansträngningar på att ta reda på vad mikroglia gör och vad som får dem att göra det. Dessa celler, visar det sig, dyker upp i musembryot på dag åtta, före någon annan hjärncell, vilket tyder på att de kan hjälpa till att styra resten av hjärnans utveckling - och kan bidra till hur många neuroutvecklingssjukdomar som helst när de går fel.
Samtidigt utökar hon också sin studie av hur olika ämnen avgör vad som händer i hjärnan. C1q är faktiskt bara det första i en serie av proteiner som ackumuleras på synapser markerade för eliminering. Stevens har börjat avslöja bevis på att det finns ett brett spektrum av skyddande don't eat me-molekyler också. Det är balansen mellan alla dessa signaler som reglerar om mikroglia kallas för att förstöra synapser. Problem hos vem som helst kan, tänkbart, förstöra systemet.
Bevis växer nu för att mikroglia är involverade i flera neuroutvecklingsproblem och psykiatriska problem. Den potentiella kopplingen till schizofreni som avslöjades i januari dök upp efter att forskare vid Broad Institute, ledd av Steven McCarroll och en doktorand vid namn Aswin Sekar, följde ett spår av genetiska ledtrådar som ledde dem direkt till Stevens arbete. Under 2009 hade tre konsortier från hela världen publicerat artiklar som jämförde DNA hos personer med och utan schizofreni. Det var Sekar som identifierade ett möjligt mönster: ju mer en specifik typ av protein fanns i synapser, desto högre risk att utveckla sjukdomen. Proteinet, C4, var nära besläktat med C1q, det som först identifierades i hjärnan av Stevens och Barres.
McCarroll visste att schizofreni drabbar sent i tonåren och tidig vuxen ålder, en tid då hjärnkretsar i den prefrontala cortex genomgår omfattande beskärning. Andra hade funnit att områden i den prefrontala cortex är bland de som drabbats mest av sjukdomen, vilket leder till massiv synapsförlust. Kan det vara så att överbeskärning av oseriösa mikroglia är en del av det som orsakar schizofreni?
För att ta reda på det tog Sekar och McCarroll kontakt med Stevens, och de två labben började hålla gemensamma veckomöten. De visade snart att C4 också hade en roll vid beskärning av synapser i hjärnan på unga möss, vilket tyder på att för höga nivåer av proteinet verkligen kan leda till överbeskärning – och till att hjärnvävnaden tunnas ut som verkar uppstå som symtom som t.ex. psykotiska episoder förvärras.
Om hjärnskadorna som ses vid Parkinsons och Alzheimers härrör från överbeskärning som kan börja tidigt i livet, varför visar sig inte symtomen på dessa sjukdomar förrän senare? Barres tror att han vet. Han konstaterar att hjärnan normalt kan kompensera för skada genom att koppla om sig själv och generera nya synapser. Den innehåller också mycket redundans. Det skulle förklara varför patienter med Parkinsons sjukdom inte visar märkbara symtom förrän de har förlorat 90 procent av nervcellerna som producerar dopamin.
Det kan också betyda att subtila symtom faktiskt kunde upptäckas mycket tidigare. Barres pekar på en studie av nunnor publicerad 2000. När forskare analyserade uppsatser som nunnorna hade skrivit när de gick in i sina kloster årtionden tidigare, fann de att kvinnor som utvecklade Alzheimers hade visat mindre idétäthet även i 20-årsåldern. Jag tror att implikationen av det är att de kan vara livslånga sjukdomar, säger Barres. Sjukdomsprocessen kan pågå i årtionden och hjärnan kompenserar bara, kopplar om, gör nya synapser. Vid någon tidpunkt utlöses mikroglia för att ta bort för många celler, hävdar Barres, och symtomen på sjukdomen börjar visa sig fullt ut.
Att förvandla denna insikt till en behandling är långt ifrån okomplicerat, för mycket är fortfarande oklart. Kanske bestäms ett alltför aggressivt svar från mikroglia av någon kombination av genetiska varianter som inte delas av alla. Stevens noterar också att sjukdomar som schizofreni inte orsakas av en mutation; snarare orsakar ett brett spektrum av mutationer med små effekter problem när de agerar tillsammans. Generna som styr produktionen av C4 och andra proteiner från immunsystemet är kanske bara en del av historien. Det kan förklara varför inte alla som har en C4-mutation kommer att fortsätta att utveckla schizofreni.
Icke desto mindre, om Barres och Stevens har rätt i att immunsystemet är en vanlig mekanism bakom förödande hjärnsjukdomar, är det i sig ett grundläggande genombrott. Eftersom vi inte har känt till mekanismerna som utlöser sådana sjukdomar, har medicinska forskare bara kunnat lindra symtomen snarare än att attackera orsakerna. Det finns inga tillgängliga läkemedel för att stoppa eller ens bromsa neurodegeneration vid sjukdomar som Alzheimers. Vissa läkemedel höjer signalsubstanser på ett sätt som kortfattat gör det lättare för individer med demens att bilda nya synaptiska kopplingar, men de minskar inte hastigheten med vilken befintliga synapser förstörs. På samma sätt finns det inga behandlingar som tar itu med orsakerna till autism eller schizofreni. Även att bromsa utvecklingen av dessa störningar skulle vara ett stort framsteg. Vi kanske äntligen går efter sjukdomar som har löpt okontrollerat i generationer.
Vi är en bit bort från ett botemedel, säger Stevens. Men vi har definitivt en väg framåt.
Adam Piore är en frilansskribent som skrev Ett chockerande sätt att fixa hjärnan i november/december 2015.